Marketingmanagement I: Übersicht

Geschätzte Lesezeit: 6 Minuten.

Ohne Kunden kein Unternehmen – diese Formel klingt simpel. Gleichzeitig fordert sie heraus, Produkte, Dienstleistungen und Unternehmen radikal aus der Kundenperspektive zu betrachten.
Diese Aufgabe, die Kundenperspektive für das Unternehmen zu analysieren, wird als Marketingmanagement bezeichnet. Diese Artikelserie will Marketingmanagement beschreiben. Dazu wird die Marketingstrategie in drei Teile zerlegt: Analyse, Strategie und Handlung. Das so entstandene Framework wird in diesem Artikel diskutiert.

Analyse: Die fünf C
- Kunde (Customer): Was sind die Wünsche und Bedürfnisse der Kunden? Wessen Bedürfnisse können gewinnbringend befriedigt werden? Wie kann Marketing einen Mehrwert für diesen Kunden schaffen und seinen Entscheidungsprozess beeinflussen? Wer ist im B2B-Kontext Teil der Entscheidung?
- Unternehmen (Company): Welche Kernkompetenzen (z.B. Ressourcen, Fachkenntnisse) hat oder braucht das Unternehmen, damit die Anforderungen des Kunden erfüllt werden können? Worin ist das Unternehmen sehr, sehr gut? Was unterscheidet das Unternehmen von anderen Firmen auf dem Markt, indem es Wert für den Kunden schafft?
- Wettbewerb (Competitor): Gibt es weitere Unternehmen, die die Bedürfnisse des Kunden erfüllen oder erfüllen wollen? Falls der Kunde nicht das eigene Produkt wählt, um sein Problem zu lösen, wessen Produkt wird stattdessen gewählt? Grundsätzlich sollen hier sowohl aktuelle als auch aufstrebende Wettbewerber einbezogen werden.
- Partner (Collaborators): Gibt es andere Unternehmen, mit denen eine Zusammenarbeit erforderlich oder hilfreich ist, um die Bedürfnisse Ihrer Kunden zu erfüllen? Wird z.B. Unterstützung bei der Herstellung benötigt? Oder muss vielleicht ein Teil oder eine Technologie eingekauft werden, damit das Produkt oder die Dienstleistung Erfolg haben?
- Umfeld (Context): Welche Umweltfaktoren begrenzen oder erweitern die Fähigkeiten des Unternehmens? Diese Faktoren können politische, wirtschaftliche, soziale / kulturelle, technologische, ökologische oder rechtliche Faktoren sein. Was passiert auf dem Markt, auf dem das Unternehmen tätig ist? Wie kann der Markt (und das Unternehmen) zu Erfolg oder Misserfolg gelenkt werden?
Strategie: STP
- Segment: Kann der Kunde in Kundengruppen unterteilt werden? Der effektivste Ansatz besteht darin, sie nach Wünschen und Bedürfnissen in Bezug auf das Produkt zu gruppieren. Die meisten Unternehmen segmentieren auch nach demografischen Merkmalen und nach psychographischen und Verhaltensunterschieden.
- Ziel (Target): Targeting ist der Prozess der Entscheidung, welches Segment zuerst mit dem Marketingplan angegangen werden soll, und ob bzw. in welcher Reihenfolge durch die verbleibenden adressierbaren Segmente vorgegangen werden soll. Dabei sollten die Stärken und Schwächen des Unternehmens, die Ziele, die benötigten Ressourcen, die verfügbaren Mitarbeiter und die möglichen finanziellen Erträge jedes Segments berücksichtigt werden.
- Position: Wie wird das Angebot den Kunden präsentiert? Positionierung beinhaltet die Entwicklung einer Positionierungsaussage, die den Zielkunden, seine Wünsche, den Produkttyp / die Produktkategorie und den Hauptnutzen des Produkts identifiziert.
Handlung: Die vier P
- Produktpolitik (Product): Durch Produkte, Dienstleistungen und Erfahrungen wird Wert für den Kunden geschaffen. In welchem Maße benötigt der Kunde ein komplettes Produkt oder eine „schlüsselfertige Komplettlösung“? Wie kann maximaler Wert für den Kunden geschaffen werden, sodass der Kunde zum Kauf bewegt wird?.
- Kommunikation (Promotion): Kommunikation macht Kunden auf das Angebot aufmerksam und baut die Marke auf, die dieses Angebot und das Unternehmen identifiziert. Hauptziel ist es, Markenbewusstsein zu schaffen und Markenwerte aufzubauen, indem Folgendes definiert wird: Markenidentität, Markenbedeutung, Markenreaktionen und Markenbeziehungen.
- Vertriebspolitik (Place): Welcher Vertriebskanal ist am besten geeignet, um den Kunden Zugang zum Produkt zu bieten? Dies kann einen einzigen exklusiven Vertriebskanal oder mehrere Kanäle (Omnichannel) umfassen, wie zum Beispiel Online-Shops, Einzelhändler oder eigene Verkaufsstellen.
- Preispolitik (Price): Wie kann der geschaffene Wert quantifiziert werden? Wie viel ist der Kunde bereit, für diesen Wert zu zahlen?
Übersicht der Beiträge
Da allein diese Einleitung bereits einige Worte enthält, wird die Detailbeschreibung dieser Punkte in einige Artikel unterteilt und dann nach und nach hier verlinkt. Derzeit sieht mein Plan so aus:
- Übersicht: Hier wird das Framework vorgestellt.
- Kundenanalyse: Der zweite Teil beginnt mit der Analyse, auf der dann Strategie und Handlung aufbauen. Besonderer Fokus liegt hierbei auf dem Kunden, welcher eines der fünf C’s darstellt.
- Strategie: Hier wird eine Strategie formuliert. Grundlage starker Strategien ist die Analyse, deren Ergebnisse regelmäßig geprüft werden müssen.
- Produktpolitik: Nun beginnt der Bereich Handlung mit dem ersten P. Begonnen wird mit Produkten, denn Entscheidungen über Produkte (und Dienstleistungen) können zur aktuellen Strategie beitragen.
- Kommunikation: Das zweite P stellt das Element des Marketings dar, das für die meisten Menschen am auffälligsten und bekannt ist: Werbung und Branding.
- Vertriebspolitik: Das dritte P der Handlungsphase wird in diesem Teil erläutert.
- Preispolitik: Das vierte P stellt die Preisstrategie dar, die den strategischen Zielen am besten entspricht. Gleichzeitig bildet dies den Abschluss der Handlungsphase, da dies das letzte verbliebene P darstellt.
- Kundenzentriertes Marketing: Es wurden Analysephase, intensive Strategie- und Handlungsphase durchlaufen. Nun folgt wieder die Analyse. In diesem letzten Teil wird all dies zum Kundenbeziehungsmanagement verbunden, denn Kunden sollen ja auch nach dem Kauf loyal sein.
- Agile Perspektive: Eine Reflektion der Theorie aus agiler Perspektive.







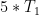 ). Auf diese Art konnte ein Großteil der Abstände von Butts et al. reproduziert und verfeinert werden.
). Auf diese Art konnte ein Großteil der Abstände von Butts et al. reproduziert und verfeinert werden.